GeneChat MCP Server
A local-first MCP server that annotates whole-genome VCF files and lets you query pharmacogenomics, disease risk, and carrier status through natural language.
README
GeneChat MCP Server
![]()
![]()
![]()
Talk to your genome.
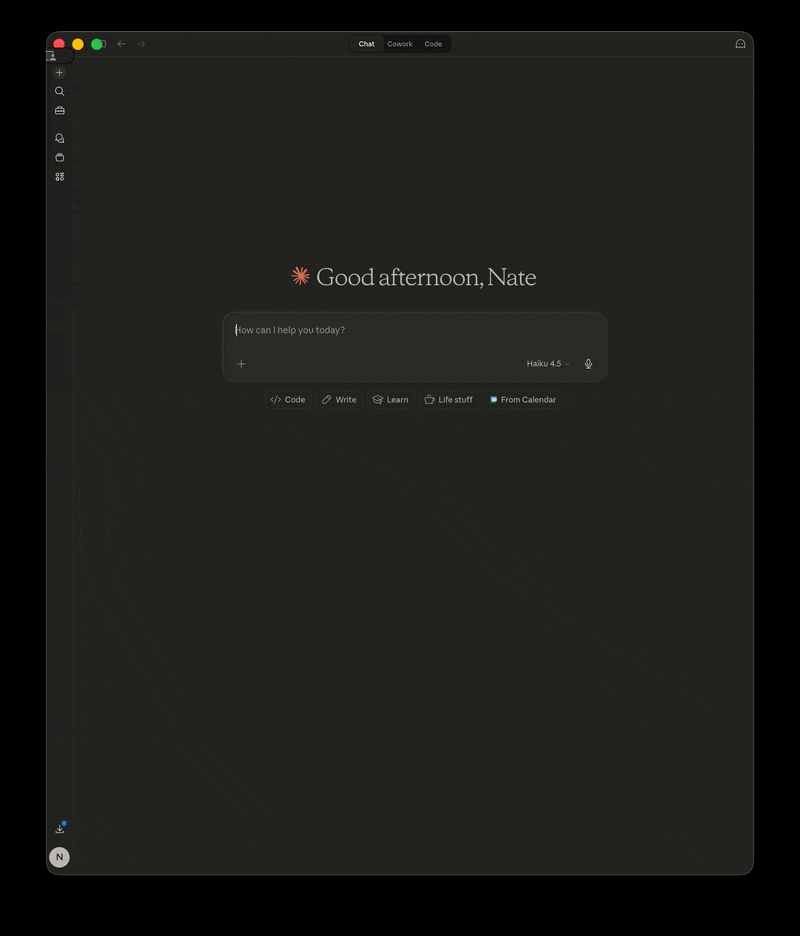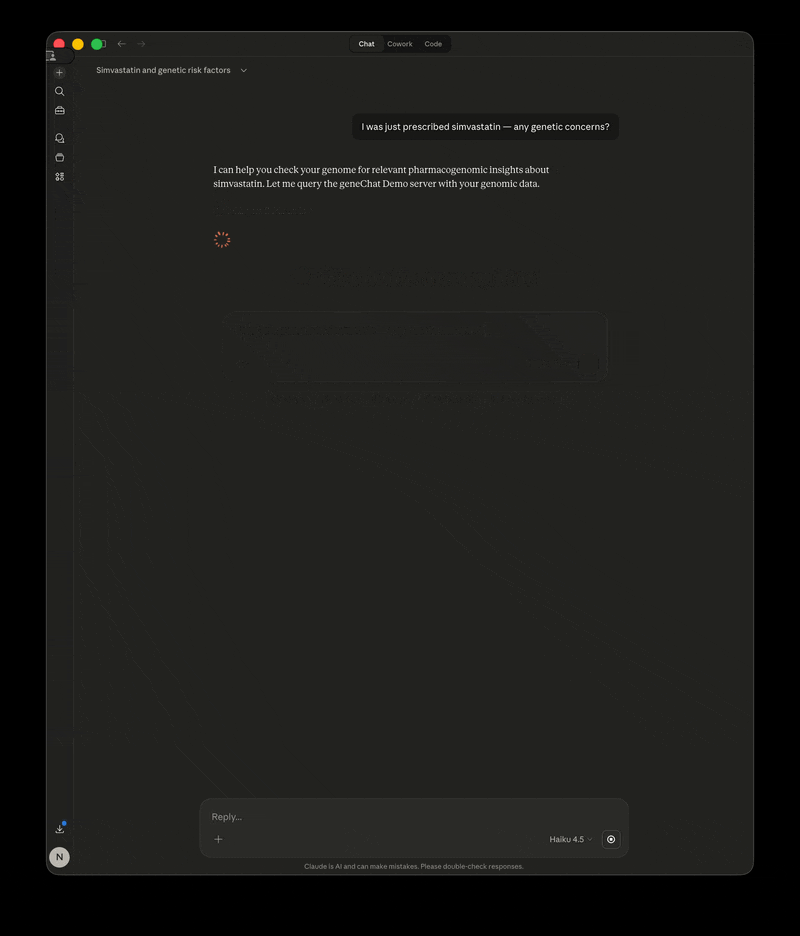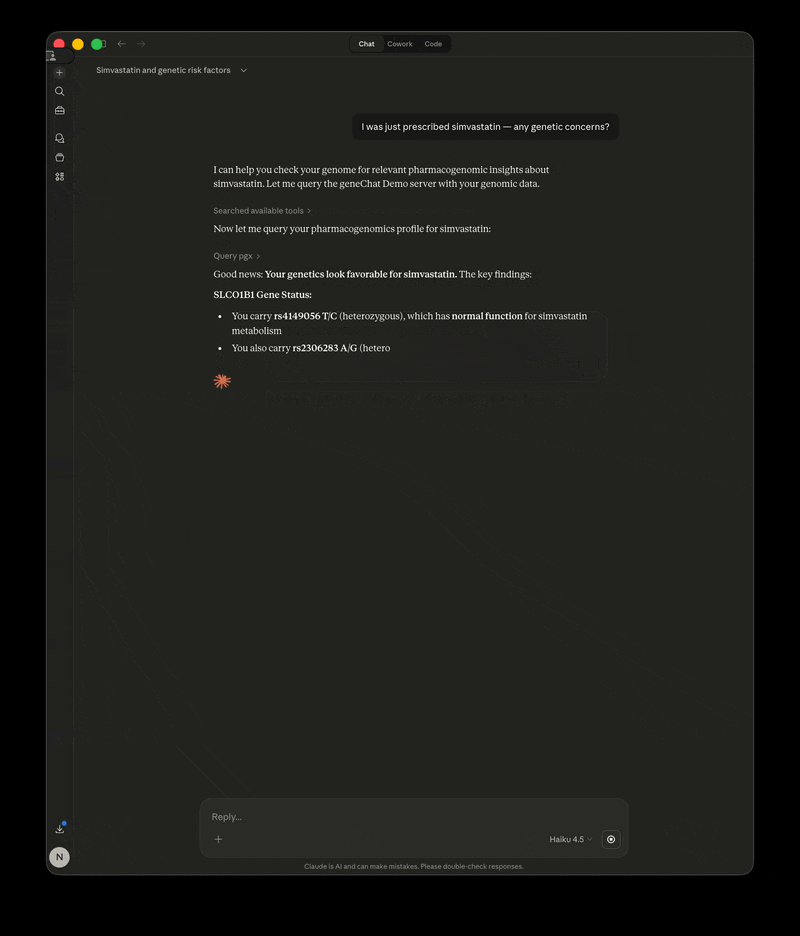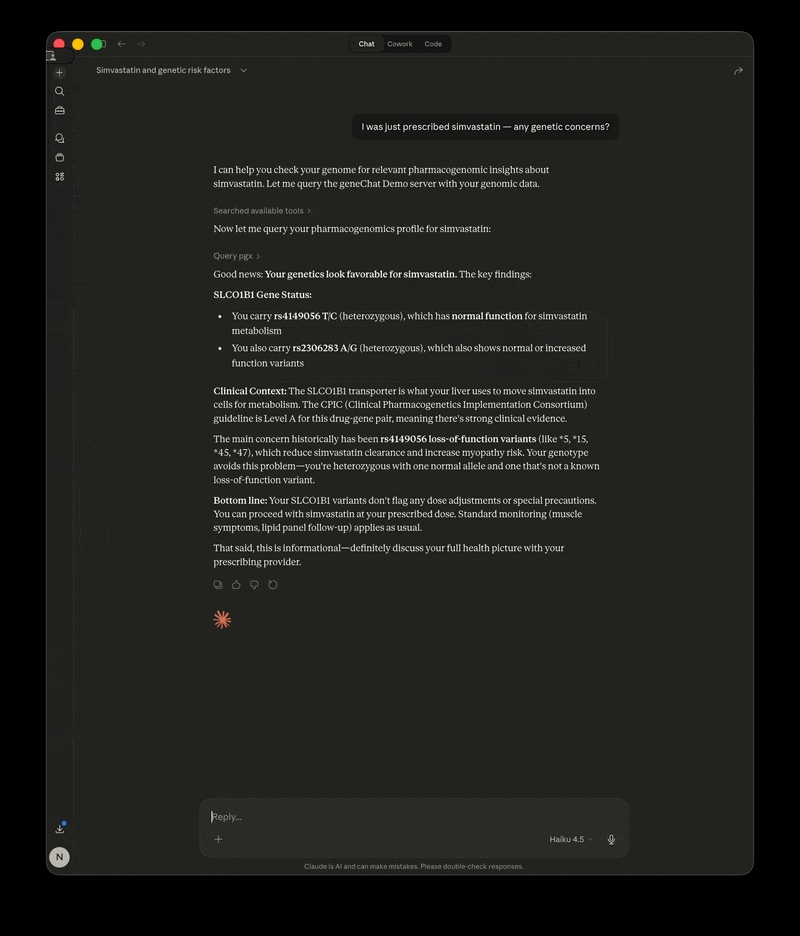
GeneChat is a local-first MCP server that annotates your whole-genome VCF once, then serves it as tools so you can ask questions about pharmacogenomics, disease risk, carrier status, and more — with your raw file never leaving your machine.
Things you can ask:
- "I was just prescribed simvastatin — any genetic concerns?"
- "I have surgery next month. What should I tell my anesthesiologist?"
- "Am I a carrier for anything concerning?"
Privacy Notice: GeneChat reads your VCF locally, but tool responses — containing your genotypes, rsIDs, and clinical interpretations — are sent to your LLM provider (e.g. Anthropic, OpenAI) as part of the conversation. Your raw VCF file is never uploaded, but the LLM does see the specific variants and findings returned by each tool call. See Security Recommendations below.
What It Does
You get your genome sequenced ($250–$900 from providers like Nucleus Genomics or Nebula Genomics). You download the raw VCF file. GeneChat annotates it once with open-source tools, then serves it locally via MCP so you can ask questions like:
- "What does my genome say about cardiovascular risk?"
- "I play soccer — any genetic injury risk factors?"
- "How should I think about my diet based on my genetics?"
The LLM calls GeneChat's tools behind the scenes, gets your specific genotypes and annotations, and interprets the results in context.
How It Works
You ask a question in your LLM → it picks the right GeneChat tool → GeneChat queries your local VCF with pysam → returns your genotype + clinical annotations → the LLM interprets the results for you.
GeneChat reads only local files and makes no network calls at runtime. However, tool responses — containing your genotypes and clinical findings — are sent to your LLM provider as part of the conversation. See Security Recommendations for details on cloud vs local LLM options.
Tools
| Tool | Purpose |
|---|---|
list_genomes |
List registered genomes so the LLM can ask which to query |
query_variant |
Look up a single variant by rsID or position |
query_variants |
Batch lookup of multiple rsIDs in a single VCF scan |
query_gene |
List notable variants in a gene with smart filter |
query_genes |
Batch query variants across multiple genes at once |
query_pgx |
Pharmacogenomics lookup by drug or gene (CPIC data) |
query_clinvar |
Find clinically significant variants |
query_gwas |
Search the GWAS Catalog by trait, gene, or variant |
calculate_prs |
Polygenic risk scores (PGS Catalog data) |
genome_summary |
High-level overview of your genome |
CLI Commands
| Command | Purpose |
|---|---|
genechat init <vcf> [--label] [--gnomad] [--dbsnp] [--gwas] [--fast] |
Full first-time setup: annotate, write config |
genechat add <vcf> [--label] |
Register a VCF file without annotation |
genechat annotate [--clinvar] [--gnomad] [--snpeff] [--dbsnp] [--all] [--stale] [--force] [--fast] [--genome] |
Build or update patch.db (auto-downloads references) |
genechat install [--gwas] [--seeds] [--force] |
Install genome-independent reference databases |
genechat status [--json] [--check-updates] |
Show all registered genomes, annotation state, and caches |
genechat licenses |
Show data source licenses for your installation |
genechat serve / genechat |
Start the MCP server |
Global flags: --version (print version), --no-color (disable colored output). Color output respects the NO_COLOR environment variable and is automatically disabled when stdout or stderr is not a TTY.
Running genechat with no subcommand in an interactive terminal shows a help summary. When stdin is piped (e.g. from an MCP client), it starts the server — so existing MCP configurations are unaffected.
Shell Completion
Enable tab completion for your shell:
genechat --install-completion
If auto-detection fails (e.g. running via uv run), specify the shell explicitly: genechat --install-completion zsh
This enables completion for subcommands, flags, and --genome labels.
Exit Codes
| Code | Meaning |
|---|---|
| 0 | Success |
| 1 | General/unexpected error |
| 2 | Invalid usage (bad arguments) |
| 3 | Configuration error (missing config, no VCF registered) |
| 4 | VCF error (file not found, invalid, missing index) |
| 5 | External tool error (bcftools/snpEff not found) |
| 6 | Network error (download failed) |
| 130 | Interrupted (Ctrl-C) |
Prerequisites
- Python 3.11+
- A consumer WGS VCF file (from Nucleus Genomics, Nebula, Sequencing.com, etc.)
- Disk for reference databases (see table below), ~2 GB for your raw VCF + patch.db
For annotation (used by genechat init and genechat annotate):
# macOS (Homebrew)
brew install bcftools brewsci/bio/snpeff
# Linux (conda)
conda install -c bioconda bcftools snpeff
VCF reading at runtime is handled by pysam, installed automatically via uv sync. No external tools needed at runtime.
Quickstart
Option A: Install from source (recommended for development)
git clone https://github.com/natecostello/genechat-mcp.git
cd genechat-mcp
uv sync
Option B: Install as a tool
uv tool install git+https://github.com/natecostello/genechat-mcp.git
# or: pip install git+https://github.com/natecostello/genechat-mcp.git
Initialize GeneChat
genechat init handles the entire setup in one command — validates your VCF, auto-fixes contig names if needed, downloads references, annotates, builds lookup tables, writes config, and prints the MCP JSON snippet:
# If installed from source:
uv run genechat init /path/to/your/raw.vcf.gz --label personal
# If installed as a tool:
genechat init /path/to/your/raw.vcf.gz --label personal
This will:
- Detect and fix bare contig names (e.g. GIAB VCFs use
1,2instead ofchr1,chr2) - Download ClinVar and SnpEff databases
- Build a patch database with functional annotations and clinical significance
- Write a
config.tomlto your OS config directory (~/Library/Application Support/genechat/on macOS,~/.config/genechat/on Linux) - Print the MCP JSON to paste into Claude Desktop or Claude Code
Optional extras (combine any flags in a single init):
# Include gnomAD population frequencies and/or GWAS trait search (~58 MB download)
uv run genechat init /path/to/your/raw.vcf.gz --gnomad --gwas
gnomAD is optional; without it, query_gene falls back to ClinVar-only filtering. GWAS enables query_gwas for trait association lookups. Both can be added after init via genechat annotate --gnomad / genechat install --gwas.
What to expect
Benchmarked on the GIAB NA12878 genome (~3.9M variants) on an 8-vCPU machine:
| Mode | Time | Peak disk | Best for |
|---|---|---|---|
Default (genechat init) |
~15 min | ~3 GB | Quick start (SnpEff + ClinVar only) |
Default + --gnomad |
several hours | ~17 GB | Low-disk machines (streams one chromosome at a time) |
--fast --gnomad --dbsnp |
~1h 45m | ~213 GB | Fast full annotation (bulk downloads, parallel workers) |
After annotation, only ~1 GB persists (VCF + patch.db). Reference databases can be deleted.
Where the time goes (--fast mode): SnpEff ~60 min (single-threaded bottleneck), gnomAD ~34 min (8 workers), dbSNP ~6 min (8 workers), ClinVar ~38s.
Don't have your genome sequenced?
You can explore GeneChat using the GIAB NA12878 benchmark genome — a well-characterized reference sample with ~3.7M variants:
# Download the benchmark VCF (~120 MB)
curl -L -O https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/NISTv4.2.1/GRCh38/HG001_GRCh38_1_22_v4.2.1_benchmark.vcf.gz
# Initialize (auto-fixes contig names, downloads references, annotates)
uv run genechat init HG001_GRCh38_1_22_v4.2.1_benchmark.vcf.gz --label giab
Then ask questions just like you would with your own genome.
Connect to your LLM
genechat init prints MCP config JSON at the end. GeneChat works with any MCP-compatible client — here are common setups:
Claude Desktop:
Copy the JSON printed by genechat init into your claude_desktop_config.json. The output varies by install method (source checkout uses uv run, tool install uses genechat directly) — paste it as-is.
Claude Code:
# Tool install (genechat on PATH):
claude mcp add genechat -- genechat
# Source install (use the command/args from genechat init output):
claude mcp add genechat -- uv run --directory /path/to/genechat-mcp genechat
Cursor / Windsurf / other MCP clients:
Add GeneChat as a stdio MCP server in your client's settings. The command is genechat (or uv run --directory /path/to/genechat-mcp genechat for source installs). Set the GENECHAT_CONFIG environment variable to your config.toml path.
Any client with an MCP config file (mcp.json, mcp_servers.json, etc.) — for source installs, paste the JSON from genechat init instead:
{
"mcpServers": {
"genechat": {
"command": "genechat",
"env": { "GENECHAT_CONFIG": "/path/to/genechat/config.toml" }
}
}
}
Try the remote demo (no installation needed):
For clients that support remote MCP servers, connect to https://genechat-demo.fly.dev/sse. In Claude Desktop, go to Settings > Connectors (requires Pro/Max/Team/Enterprise), or use the mcp-remote bridge:
{
"mcpServers": {
"genechat-demo": {
"command": "npx",
"args": ["-y", "mcp-remote", "https://genechat-demo.fly.dev/sse"]
}
}
}
Claude Code:
claude mcp add genechat-demo --transport sse https://genechat-demo.fly.dev/sse
Multiple genomes
GeneChat supports named genomes for side-by-side comparison:
# Register a second genome
uv run genechat init /path/to/partner.vcf.gz --label partner
# Check what's registered
uv run genechat status
The LLM can then query both genomes using the genome parameter on any tool and the genome2 parameter on most tools for side-by-side comparison.
Architecture
Your raw VCF is never modified. genechat init downloads reference databases (ClinVar, SnpEff, optionally gnomAD/dbSNP) once, annotates your VCF, and stores the results in a separate SQLite patch database (patch.db). At runtime, the MCP server reads your raw VCF and patch.db locally — no network calls, no external tools. Tool responses flow back to the LLM client via the MCP protocol.
Annotation Pipeline (one-time, handled by genechat init)
| Tool | What it adds | Install |
|---|---|---|
| SnpEff | Functional annotation — gene name, effect type, impact level, protein change | brew install brewsci/bio/snpeff |
| bcftools | Database annotation — transfers ClinVar/gnomAD/dbSNP fields into patch.db | brew install bcftools |
For incremental updates of individual annotation layers (e.g., updating ClinVar without re-running the full pipeline), see docs/annotation-updates.md.
Reference Databases
| Database | What it provides | Size | Flag |
|---|---|---|---|
| ClinVar | Clinical significance, disease/condition name, review status | ~100 MB | Default |
| SnpEff DB | Gene/transcript models for functional impact prediction | ~1.6 GB | Default |
| gnomAD | Population allele frequencies (global + per-population) | ~150 GB | --gnomad |
| dbSNP | rsID identifiers for each genomic position | ~20 GB | --dbsnp |
| GWAS Catalog | 1M+ genome-wide association study findings | ~58 MB download, ~300 MB on disk | --gwas |
Default genechat init downloads ClinVar + SnpEff (~2 GB). Optional annotation layers are enabled with flags (e.g. genechat annotate --gnomad). Genome-independent databases like GWAS are installed separately (genechat install --gwas).
Seed Data Pipeline
Gene coordinates, PGx guidelines, and PRS weights are pre-built from external APIs. The source repo contains the raw TSVs in data/seed/; pip-installed users get the prebuilt SQLite lookup_tables.db directly. Either way, genechat init ensures the database is ready — no manual steps needed.
To refresh seed data from upstream APIs (fetches latest from HGNC, Ensembl, CPIC, and PGS Catalog):
genechat install --seeds
Runtime Dependencies
At runtime, GeneChat uses only local files — no external tools, no network calls. (Tool responses are returned to the LLM client, which forwards them to the LLM provider — see Security Recommendations.)
| Library | What it does |
|---|---|
| pysam | Reads your raw VCF via tabix index |
| mcp | Implements the MCP server protocol |
| SQLite (stdlib) | Queries lookup tables for gene coordinates, drug info, PRS weights |
| pydantic | Validates tool inputs and config |
| typer | CLI framework (subcommands, flags, shell completion) |
| platformdirs | OS-standard config and data directories |
Data Source Licenses
License obligations depend on which annotation layers you install. The base install includes bundled seed data (PGx, PRS, warning genes) using public-domain, CC0, CC BY 4.0, and HPO-licensed data — citations are appreciated. genechat install --seeds refreshes this data from upstream APIs.
| Install path | Sources | License | Key obligation |
|---|---|---|---|
Default (genechat init) |
ClinVar, SnpEff, CPIC, HGNC, Ensembl | Public domain / MIT / CC0 | None (citation appreciated) |
--gnomad |
gnomAD | ODbL 1.0 | Attribution required; share-alike on derivative databases |
--dbsnp |
dbSNP | Public domain | None |
--gwas |
GWAS Catalog | CC0 | None |
| Bundled (PRS) | PGS Catalog | EBI ToU + per-score | Cite catalog paper + individual score publications |
| Bundled (warnings) | HPO, ACMG SF | Custom / academic | HPO: cite, show version, do not modify; ACMG: cite |
Run genechat licenses to see which licenses apply to your specific installation. See docs/licenses.md for full attribution text, citation DOIs, and the gnomAD ODbL produced-works distinction.
Security Recommendations
| Data | Where it lives | Transmitted? |
|---|---|---|
| Your VCF file | Your machine only | Never |
| Tool responses (genotypes, rsIDs, findings) | Sent to LLM provider per tool call | Yes |
| Conversation history | MCP client logs (local) | Depends on client settings |
GeneChat makes zero network calls at runtime. However, every tool response — containing your genotypes, rsIDs, and clinical interpretations — is returned to the LLM as part of the conversation.
With a cloud LLM (Claude, ChatGPT, etc.): your raw VCF stays local, but tool responses are sent to the provider's servers. The provider's data policies apply to this content.
With a local/self-hosted model (Ollama, llama.cpp, etc.): everything stays on your machine. If you want maximum privacy, use an MCP client configured to run a local model so tool responses never leave your machine.
Store your VCF on an encrypted volume and chmod 600 your VCF and config files. genechat init sets restrictive permissions on the config automatically. MCP clients may log conversation history locally — be aware of cloud sync on those directories. See docs/security.md for platform-specific encryption instructions (APFS, LUKS).
Privacy summary: No telemetry, no analytics, no data collection. Your raw VCF file never leaves your machine. Tool responses containing your genetic findings are sent to your LLM provider as part of the conversation — use a local LLM if this is a concern.
Development / Testing
uv sync --extra dev
uv run pytest
uv run ruff check . && uv run ruff format --check .
The test VCF (tests/data/test_sample.vcf.gz) is auto-generated by a pytest fixture on first run.
End-to-End Testing with GIAB NA12878
Optional e2e tests against the GIAB NA12878 benchmark genome (~3.7M variants):
# Download and init GIAB (see "Don't have your genome sequenced?" above)
# Include --dbsnp for rsID-based lookups in e2e tests
uv run genechat init HG001_GRCh38_1_22_v4.2.1_benchmark.vcf.gz --label giab --dbsnp
# Run e2e tests (point to the chrfixed VCF if contig rename was applied):
export GENECHAT_GIAB_VCF=./HG001_GRCh38_1_22_v4.2.1_benchmark_chrfixed.vcf.gz
uv run pytest tests/e2e/ -v
# Fast only (skip full-VCF scans):
uv run pytest tests/e2e/ -v -m "not slow"
E2e tests are automatically skipped when GENECHAT_GIAB_VCF is not set.
Troubleshooting
Missing VCF index (.tbi): tabix -p vcf /path/to/your/raw.vcf.gz
Wrong genome build: GeneChat expects GRCh38 with chr prefixed chromosomes. GRCh37/hg19 VCFs need liftover first.
Missing lookup_tables.db: The lookup database ships with the package and is built automatically by genechat init. If somehow missing, rebuild with genechat install --seeds.
pysam installation issues on macOS: xcode-select --install
Verifying LLM Interpretations
GeneChat's tool responses are derived from curated databases (ClinVar, CPIC, gnomAD, PGS Catalog) and your local VCF. Results may be incomplete due to query limits, missing annotations, or database version differences — always verify against primary sources. The LLM interpreting that data can also hallucinate — inventing clinical significance, overstating risk, fabricating study references, or filling in gaps with training-data knowledge that may be outdated or wrong.
Practices that reduce this risk:
-
Require citations. Ask the LLM to cite specific sources (PMIDs, ClinVar accession numbers, CPIC guideline URLs) for every clinical assertion. If it can't point to a source from the tool response, treat the claim with skepticism.
-
Spot-check against primary sources. Any assertion about a specific variant can be verified in seconds:
-
Cross-model verification. For high-stakes findings, run the same query through a second LLM. Agreement on specific claims (citing the same guideline or variant classification) increases confidence. Disagreement is a signal to check primary sources.
-
Distinguish tool data from LLM synthesis. GeneChat tool responses are clearly structured (genotype, annotation, clinical significance, population frequency). Anything the LLM adds beyond that — dietary recommendations, risk quantification, drug dosing suggestions — is LLM-generated interpretation, not database fact.
-
Be wary of confident gap-filling. If GeneChat returns "no ClinVar entry found" for a variant, but the LLM still offers a clinical interpretation, that interpretation came from training data, not from your annotated genome.
Important Disclaimer
GeneChat is an informational tool, not a medical device. It is not a substitute for professional genetic counseling or medical advice. Always discuss genetic findings with a qualified healthcare provider before making health decisions.
License
MIT
Recommended Servers
playwright-mcp
A Model Context Protocol server that enables LLMs to interact with web pages through structured accessibility snapshots without requiring vision models or screenshots.
Magic Component Platform (MCP)
An AI-powered tool that generates modern UI components from natural language descriptions, integrating with popular IDEs to streamline UI development workflow.
Audiense Insights MCP Server
Enables interaction with Audiense Insights accounts via the Model Context Protocol, facilitating the extraction and analysis of marketing insights and audience data including demographics, behavior, and influencer engagement.
VeyraX MCP
Single MCP tool to connect all your favorite tools: Gmail, Calendar and 40 more.
graphlit-mcp-server
The Model Context Protocol (MCP) Server enables integration between MCP clients and the Graphlit service. Ingest anything from Slack to Gmail to podcast feeds, in addition to web crawling, into a Graphlit project - and then retrieve relevant contents from the MCP client.
Kagi MCP Server
An MCP server that integrates Kagi search capabilities with Claude AI, enabling Claude to perform real-time web searches when answering questions that require up-to-date information.
E2B
Using MCP to run code via e2b.
Neon Database
MCP server for interacting with Neon Management API and databases
Exa Search
A Model Context Protocol (MCP) server lets AI assistants like Claude use the Exa AI Search API for web searches. This setup allows AI models to get real-time web information in a safe and controlled way.
Qdrant Server
This repository is an example of how to create a MCP server for Qdrant, a vector search engine.